«12 месяцев». Прогрессируюшая оссифицирующая фибродисплазия. Часть первая: дузеры и фрэгглы нашего скелета
Информационно-просветительский гуманитарный проект «12 месяцев» — это цикл материалов о необычных людях – пациентах с редкими (орфанными) болезнями, о которых не написано в студенческих учебниках. Считается, что вероятность встретить на профессиональном пути редкого пациента у обычного врача ничтожно мала, поэтому в академических аудиториях им не уделяют должного внимания, что в повседневной жизни приводит к диагностическим ошибкам, упущенному времени и поломанными судьбами и жизням. Проект «12 месяцев» реализуют студенты и ординаторы – будущие и нынешние специалисты, активно изучающие генетические методы диагностики, их место в современной врачебной работе. Материалы готовятся на кафедре патологической анатомии СЗГМУ им. И.И. Мечникова (Санкт-Петербург) при поддержке научно-практического журнала «Гены и Клетки», группы компаний ИСКЧ, блога истории медицины и порталов Indicator.Ru и «Нейроновости». Каждый проект будет состоять из трех материалов: рассказа о заболевании с видеотаймлайном его изучения, и пациентской истории. Научные редакторы проекта — Алексей Паевский и Роман Деев. Восьмой цикл статей посвящен фибродисплазии оссифицирующей прогрессирующей (ФОП).

Ставь лайк, если помнишь кто такие дузеры!
В «интернетах» тех, кто лайканул назвали бы олдами. дузеры – герои детского телевизионного сериала «Скала фрэгглов» из 80-х годов прошлого века… Все серии они как заговоренные непрерывно возводили строительные конструкции, а фрэгглы – существа, населявшие пещеры, осознанно или неосознанно непрерывно рушили постройки (они их попросту ели!). Хрупкий баланс строительства и разрешения сохранял комфортную обстановку в микромире сказочных существ.
В микромире нашего скелета есть свои дузеры – это клетки «остеобласты» - они строят костное вещество – минерализованный каркасный эндоскелет. И есть свои фрэгглы – остеокласты, они разрушают постройки костных дузеров. Баланс между этими процессами гарантирует нам надежность скелета. А теперь представьте, что фрэгглы разбушевались, и разрушают больше, чем могут построить остеобласты – костное вещество скелета утрачивается, скелет становится хрупким. Или противоположный процесс – дузеры вышли из под контроля и стали застраивать костными балками все доступное им пространство – так костного вещества становится слишком много, много настолько, что оно мешает проходящим рядом сосудам и нервам, вытесняет костный мозг – такое заболевание называется остеопетроз – болезнь «каменных костей», это генетическое заболевание, в основе которого нарушение в генах, управляющих нашими внутренними дузерами.

Но бывает и того хуже – дузеры выходят за флажки, за пределы скелета и начинают стройку кости там, где ей делать совершенно нечего. Сегодняшний наш рассказ о «каменных людях» - пациентах, страдающих особой генетической болезнью – фибродисплазией оссифицирующей прогрессирующей (ФОП).
Среди редких (орфанных) заболеваний, безусловно, имеются свои особенные антирекордсмены. Одной из таких болезней является фибродисплазия оссифицирующая прогрессирующая (сокращенно – ФОП/ПОФ). В среднем по миру она встречается с частотой 1 случай на 2 миллиона человек [1, 2]. В Российской Федерации таких больных насчитывается около 70. Фибродисплазия оссифицирующая прогрессирующая (ФОП) – генетическое нарушение, характеризующееся «окостенением» мышц, сухожилий, связок, фасций и подкожных тканей – т.е. развитием костей в «мягких тканях» [3].

ФОП – тяжелое, инвалидизирующее и постоянно прогрессирующее (отсюда и название) заболевание, значительно снижающее качество и длительность жизни больных. При своевременной диагностике и лечении средняя продолжительность жизни больных составляет около 40-60 лет [4, 5].
В чём проблема?
История описания ФОП начинается с конца XVII века, когда французский врач Гай Пэтин в 1692 году в письме своему коллеге описывает пациентку с ФОП: «Сегодня я увидел женщину, которая стала твёрдой, как дерево». Затем в 1736 году английский хирург Джон Фрике обращается в Лондонское королевское общество с описанием необычного случая: «Пришёл с виду здоровый мальчик 14 лет с единственным вопросом: что нужно сделать, чтобы исцелить его от множества больших опухолей на спине? Они возникали из рёбер, всех позвонков, шеи и доходили вплоть до крестца, по форме напоминали разветвления коралла, образовывая что-то в виде корсета. Поэтому это заболевание также называют «болезнью второго скелета».
Что же лежит в основе процесса, результатом которого является формирование «скелета в скелете», подобно панцирю черепах? ФОП известна человечеству с конца XVII века, однако более подробное изучение её причин и механизмов началось лишь в конце прошлого столетия и связано оно с деятельностью двух американских врачей – Фредерика Каплана и Майкла Заслоффа. В недалёком 1989 году они открыли исследовательский проект по изучению ФОП на базе медицинского университета Пенсильвании, впоследствии там появилась и молекулярная лаборатория. В 2006 году им удалось найти единственную и достоверную причину возникновения этой болезни – ей оказалась всего лишь единичная мутация на 2-й хромосоме в гене рецептора активина I типа - ACVR1, ответственного за образование трансмембранного рецепторного белка Alk2 [6].

Дело в том, что вся генетическая информация (ДНК) в организме человека записана в виде отдельных нуклеотидов (подобно буквам алфавита) – аденин (А), тимин (Т), цитозин (С) и гуанин (G). Двуцепочечная ДНК является матрицей в процессе транскрипции для образования цепочки нуклеотидов РНК. Она отличается от ДНК тем, что нуклеотид тимин (Т) заменяется на урацил (U). РНК направляется к рибосомам для преобразования нуклеотидов в конкретные аминокислоты – это процесс трансляции. При этом каждой тройке нуклеотидов (триплет) соответствует лишь единственная аминокислота. Впоследствии аминокислоты формируют белок, который выполняет генетически возложенные на него функции. В случае с ФОП мутация в гене ACVR1 влечёт за собой замену нуклеотида гуанина (G) на аденин (A), из-за чего аминокислота аргинин меняется на гистидин. В результате получается другой по форме рецепторный белок Alk2, способствующий образованию костной ткани в нетипичных местах.
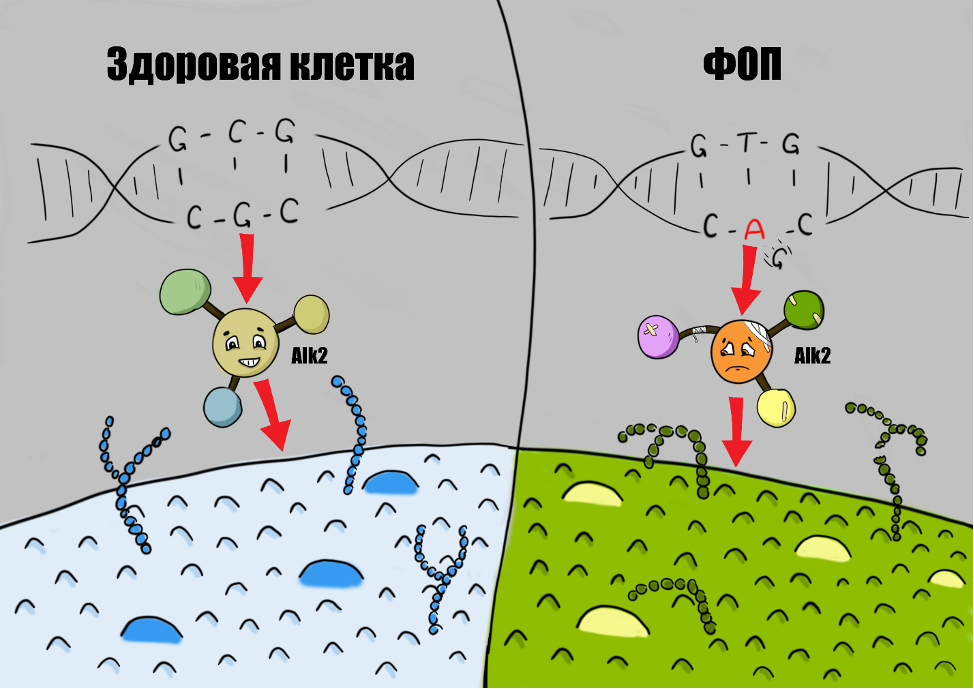
В норме рецептор Alk2 реагирует только на специальные белки (лиганды) группы костного морфогенетического белка (Bone morphogenetic protein, BMP) и белка Activin A – именно они отвечают за образование костной ткани в процессе развития человека через множество сигнальных путей.
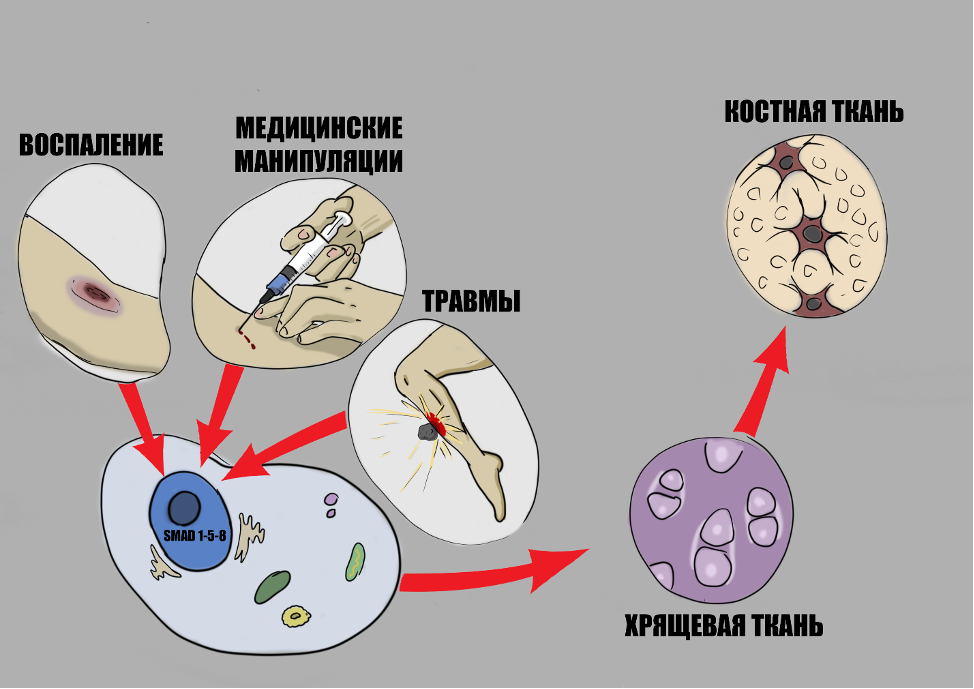
В результате того, что помимо здорового рецептора образуется и мутантный трансмембранный белок Alk2, в ответ на триггерное воздействие (травма, воспаление, медицинские манипуляции и т.п.) увеличивается уровень костного морфогенетического белка, происходит избыточная активация рецептора и он активизируется даже в отсутствии лигандов [7]. При этом иммунные клетки (макрофаги, лимфоциты и др.) также играют важную роль – являясь главными участниками воспаления, они выделяют многие биологически активные вещества, в том числе стимулирующие образование костной ткани [8].
Затем через сигнальные пути SMAD 1/5/8 в ядре стволовых стромальных клеток, расположенных в мыщцах, сухожилиях, связках и фасциях активируются гены, ответственные за последовательное образование хрящевой и затем уже костной тканей на месте повреждения. Вышеупомянутые механизмы являются ключевыми, однако не единственными, многие молекулярные механизмы остаются неизвестными.
При этом описаны и другие более редкие мутации, они варьируют по своим проявлениям, однако общим для них является наличие мутации в гене ACVR1.
Пациенты с ФОП рождаются внешне абсолютно здоровыми. Одним из ранних патогномоничных (специфических) признаков является деформация большого пальца стопы – он короткий и вогнут кнутри [9]. В течение первого десятилетия жизни у детей с ФОП появляются болезненные припухлости мягких тканей (так называемые вспышки ФОП). Они могут появляться как вследствие травм, так и спонтанно без видимой причины. В связи с редкостью ФОП, зачастую врачи путают эти проявления с опухолями, что приводит к лишним вмешательствам и усугублению состояния.
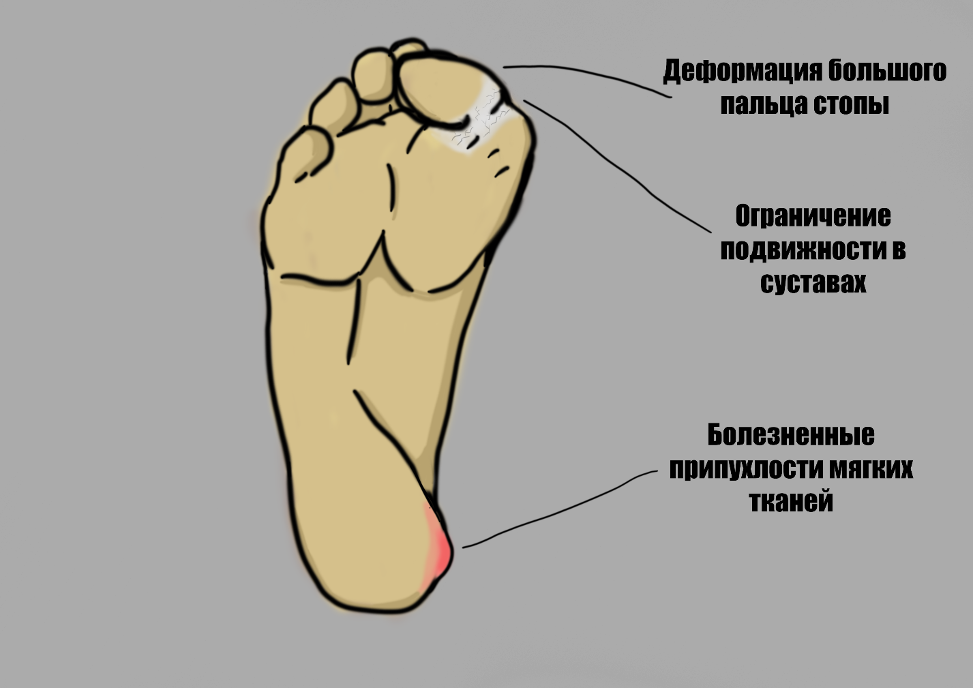
В результате травм, падений, инъекций и других вмешательств в сформированных припухлостях происходит процесс развития кости – остеогенез, приводящий к ограничению подвижности в суставах. Через 10 лет после манифестации заболевания наблюдается ограничение движений в позвоночнике и плечах, через 20 лет – в одном или обеих бёдрах. К 30 годам большинство пациентов прикованы к инвалидному креслу и неспособны самостоятельно себя обслуживать [10]. Больные с ФОП вынуждены ограничивать себя во многих повседневных занятиях, дети с ФОП зачастую находятся на домашнем обучении и под пристальным надзором родителей. В основном «вспышки» остеогенеза локализуются в шее, позвоночнике, плечах, затем поражаются предплечья, тазовая область и коленные суставы. Следует отметить, что при этом е описано вовлечение в патологический процесс диафрагмы, сердца, мышц языка и глаз. Иногда «вспышки» могут возникать без явной на то причины, их длительность варьирует от 1-2 недель до месяцев, при этом они сопровождаются сильной болью и скованностью движений.
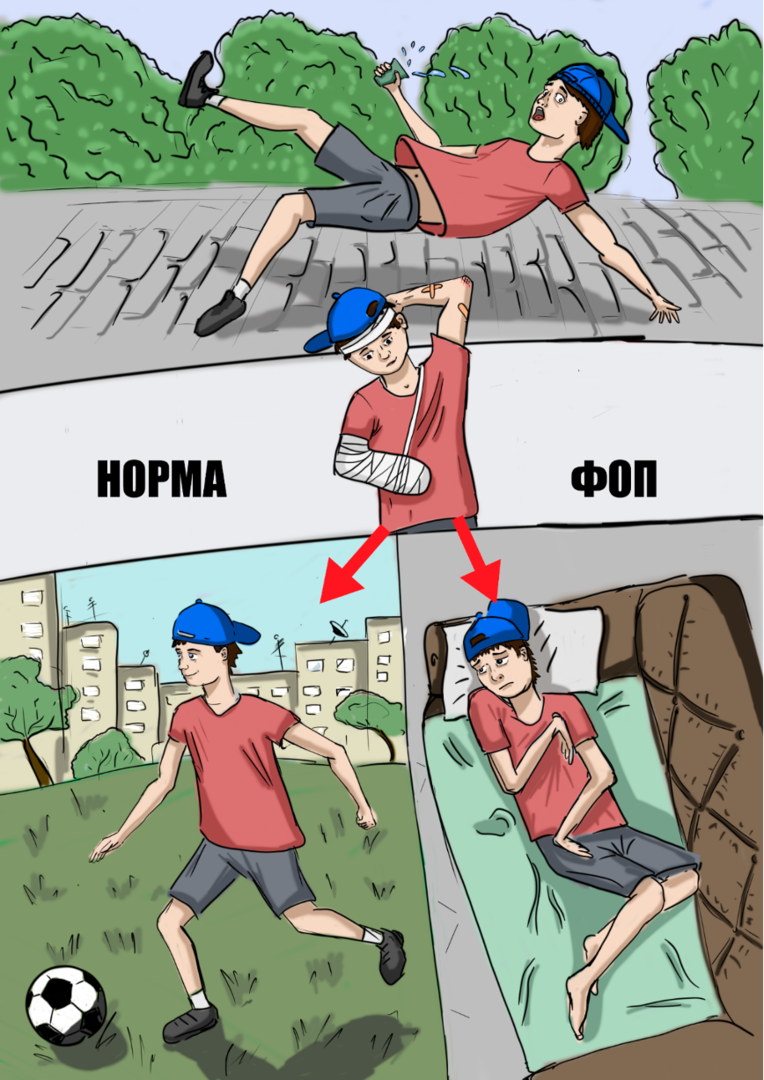
Несмотря на то, что при ФОП главная дыхательная мышца – диафрагма не поражается, больные нередко погибают от дыхательной недостаточности. Это связано с возникновением «синдрома торакальной недостаточности» - из-за грубых деформаций и искривлений грудной клетки лёгкие неспособны расправляться, они оказываются «замурованы» в собственной грудной клетке. Присоединение пневмонии на фоне ограниченного движения грудной клетки значительно усугубляет состояние пациента.
Как подтвердить заболевание?
Как мы выяснили, с самого рождения ФОП заподозрить достаточно тяжело. Помимо неявной клинической картины, важное место в диагностике ФОП занимает молекулярная диагностика – проверка гена ACVR1 и выявление в нем частой мутации, что точно подтверждает наличие заболевания.
Как лечить?
В настоящее время ФОП – тяжелое, неуклонно прогрессирующее заболевание без специфического лечения. В рутинной практике в основном используются препараты для патогенетической терапии, воздействующие не на причину заболевания, а на отдельные его механизмы. Например, для предотвращения образования новой кости после травм применяются НПВС (аспирин, целекоксиб и т.п) и глюкокортикостероиды (в основном, преднизолон) [11]. Однако это не всегда подавляет остеогенез и влечёт за собой побочные эффекты от длительного приёма (к примеру, недостаточность коры надпочечников при длительном приёме глюкокортикостероидов, развитие остеопороза, синдрома Кушинга и другие).
В мире активно ведутся исследования по внедрению новых лекарственных средств для лечения ФОП. К примеру, препарат Palovaroten (Ipsen Inc.) является агонистом (активатором) RAR-гамма рецепторов, что снижает образование хрящевой ткани (следовательно, и костной в дальнейшем) [12]. Препарат успешно прошёл I (изучение у здоровых людей) и II (изучение у пациентов с ФОП) фазы исследования, сейчас препарат находится в III фазе клинических исследований. Регуляторные органы Канады 24 января 2022 года уже одобрили Palovaroten для постоянного применения пациентам с ФОП.
Существуют и другие лекарственные средства, используемые при лечении ФОП, однако большинство из них направлены на одну из цепочек механизма, но не на саму причину заболевания. В 2020 году всемирная ассоциация ФОП - ifopa.org, профинансировала первый исследовательский грант «В погоне за исцелением», направленный на реализацию лечения ФОП с генетической точки зрения [13]. Международная научная группа пока не опубликовала промежуточные результаты своих работ, но выделила несколько подходов для разработки потенциального лечения. Они основаны на внедрении аденоассоциированного вируса (AAV), который способен на:
● Gene addition - добавление нового, здорового и функционально активного гена.
● Gene silencing - подавление мутантного гена. Этот феномен реализуется посредством метода РНК-интерференции — введение специфичной молекулы РНК, которая комплементарна (соответствует) молекулам ДНК и РНК искомого мутантного гена, что влечёт за собой их дальнейшее разрушение.
● Gene replacement – замещение гена, основанное на доставке пациентам работающей копии терапевтического гена.
● Gene editing – генное редактирование, направленное на исправление имеющейся в поврежденном гене мутации. Наиболее популярной системой для редактирования генов является CRISPR/CAS9. Аденоассоциированный вектор играет роль курьера и доставляет генную конструкцию, которая «ремонтирует» поврежденный участок гена.
Куда обращаться за помощью?
● В мире существует международная ассоциация ФОП — ifopa.org.
● В Российской Федерации действует межрегиональная общественная организация «Живущие с ФОП» — foprussia.ru.
Для консультации по вопросам ФОП можно обратиться в один из трёх центров, изучающих и оказывающих лечение больным ФОП:
● Федеральное государственное бюджетное научное учреждение «Научно-исследовательский институт ревматологии им. В.А. Насоновой», https://rheumatolog.su.
● Федеральное государственное бюджетное научное учреждение «Медико-генетический научный центр им. академика Н.П. Бочкова», https://med-gen.ru.
Текст: Павел Подлужный, Роман Деев
Иллюстрации: Владислав Ефремов
Литература: 1. Connor D.M., Evans D.A. Genetic aspects of fibrodysplasia ossificans progressiva. J. Med. Genet. 1982; 19(1): 35–9. 2. Morales-Piga A., Bachiller-Corral J., Trujillo-Tiebas M.J. et al. Fibrodysplasia ossificans progressiva in Spain: epidemiological, clinical, and genetic aspects. Bone 2012; 51(4): 748–55. 3. Коваленко-Клычкова Н.А., Клычкова И.Ю., Кенис В.М. и др. Прогрессирующая оссифицирующая фибродисплазия у детей (обзор литературы и анализ 5 клинических случаев). Травматология и ортопедия России 2014; 1(71). 4. Sharique M., Siddiqui Y.S., Abbas M., et al. Fibrodysplasia ossificans progressiva - a case report with brief literature review. MOJ Orthop. Rheumatol. 2020; 12(6): 132‒5. 5. Garde D. Their tissue turns to bone. Their joints freeze in place. And, finally, their hopes for treatment may be realized. https://www.statnews.com/2019/03/20/fibrodysplasia-ossificans-progressiva-fop-treatment. 6. Shore E., Xu M., Feldman G. et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006; 38: 525–7. 7. Cappato S., Giacopelli F., Ravazzolo R. et al. The Horizon of a Therapy for Rare Genetic Diseases: A "Druggable" Future for Fibrodysplasia Ossificans Progressiva. Int. J. Mol. Sci. 2018; 19(4): 989. 8. Convente M.R., Wang H., Pignolo R.J. et al. The immunological contribution to heterotopic ossification disorders. Curr. Osteoporos Rep. 2015; 13(2): 116-24. 9. Kaplan F.S, Xu M., Seemann P. et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009; 30(3): 379-90. 10. Martelli A., Santos A.R. Cellular and morphological aspects of fibrodysplasia ossificans progressiva. Lessons of formation, repair, and bone bioengineering. Organogenesis 2014; 10(3): 303-11. 11. Kaplan F.S. et al. The medical management of fibrodysplasia ossificans progressiva: current treatment considerations. Proc. Intl. Clin. Council FOP. 2019; 1-111. 12. Chakkalakal S.A., Uchibe K., Convente M.R. et al. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice with the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016; 31(9): 1666-75. 13. https://www.ifopa.org/curefop_get_facts.